Forscher entwickeln neue Therapie für besonders aggressive Tumorform
Eine seltene Form von Weichteiltumor lässt sich schwer einordnen und behandeln. Eine neue Studie fand in den Krebszellen Hinweise auf passendere Therapien.
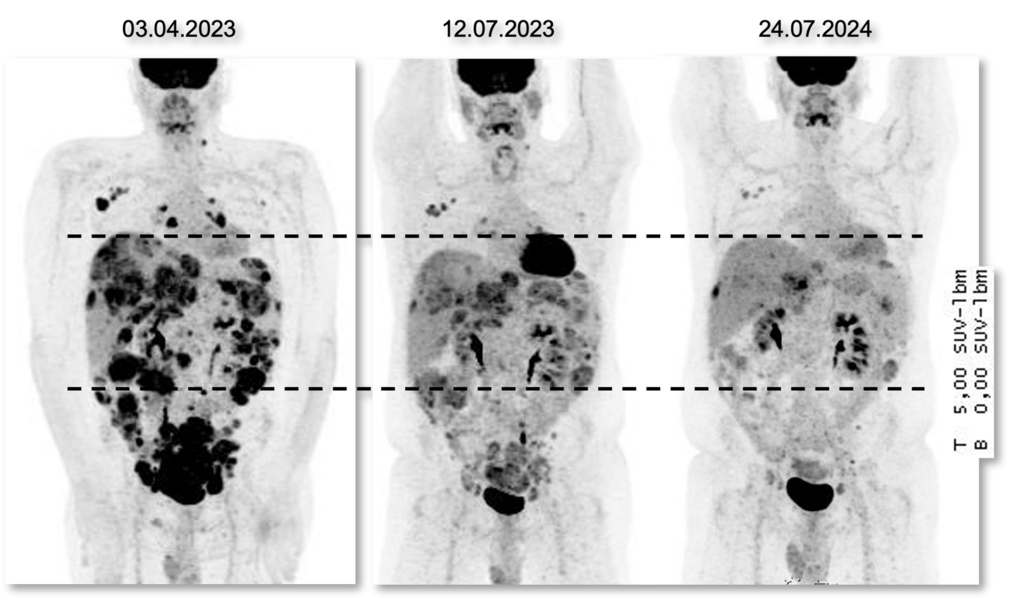
PET-Aufnahmen (Positronen-Emissions-Tomografie) eines Patienten zu drei Zeitpunkten zeigen ein sehr gutes Ansprechen auf das ADC Trastuzumab-Deruxtecan, das ungefähr 18 Monate angehalten hat. © NCT Heidelberg
Ein Weichteiltumor entsteht in Geweben wie Muskeln, Fett oder Bindegewebe. Eine besonders aggressive Form davon wird in einer aktuellen Heidelberger Studie besprochen. Er ist zwar sehr selten, betrifft aber oft junge Menschen und lässt sich nur schwer behandeln. Die Diagnose fällt häufig schwer, die Therapiemöglichkeiten sind begrenzt. Die Arbeit im Fachjournal Nature Communications zeigt nun: Bei 27 Prozent der Patienten wurde der Tumor erst durch eine genaue Analyse der Krebszellen richtig erkannt, und bei 62 Prozent wirkten individuell angepasste Therapien messbar.
Forscher vom Nationales Centrum für Tumorerkrankungen (NCT) Heidelberg untersuchten 30 Patienten mit dem sogenannten desmoplastischen klein- und rundzelligen Tumor, kurz DSRCT. Der Altersdurchschnitt lag bei 30 Jahren, 26 der 30 Betroffenen waren Männer. Pro Jahr erkranken nur etwa 0,2 Menschen pro eine Million Einwohner an der seltenen Krankheit. Die Prognose ist schlecht – viele Betroffene sterben innerhalb von drei Jahren nach der Diagnose.
Seltener Weichteiltumor wird oft spät erkannt und falsch eingeordnet
Das größte Problem entsteht bereits zu Beginn der Erkrankung: In acht von 30 Fällen wurde der Tumor zunächst nicht korrekt eingeordnet. Erst eine molekulare Untersuchung brachte die richtige Diagnose. Damit betrifft das mehr als jeden vierten Patienten.
Bis zur korrekten Diagnose verging im Schnitt rund ein Jahr. In dieser Zeit erhielten einige Betroffene Therapien, die nicht zu ihrer tatsächlichen Erkrankung passten. Die ursprünglichen Einschätzungen reichten von Tumoren ohne bekannten Ursprung bis hin zu anderen Sarkomformen.
Die Unsicherheit liegt daran, dass DSRCT sehr ähnlich zu anderen Krebsarten ist. Unter dem Mikroskop wirkt der Tumor oft unauffällig und lässt sich schwer unterscheiden. Erst spezielle Analysen im Erbgut liefern klare Hinweise. Dabei wird vor allem eine bestimmte Genverbindung untersucht, die bei dieser Tumorart fast immer vorkommt und schließlich die Diagnose absichert.
Ärzteteams prüfen Daten gemeinsam und schlagen Therapien vor
Nach der Analyse besprechen Fachärzte die Ergebnisse gemeinsam in einem sogenannten Tumorboard. Dabei kommen Onkologen, Pathologen und weitere Spezialisten zusammen und bewerten jeden Fall einzeln. Auf dieser Basis entstehen Ansätze für die weitere Behandlung.
Für 28 der 30 Patienten konnten solche Empfehlungen formuliert werden. Insgesamt kamen 107 Vorschläge zusammen, die sich an den jeweiligen Eigenschaften des Tumors orientierten. Viele Vorschläge richteten sich gegen Eiweiße oder Signalwege, die das Wachstum der Krebszellen antreiben.
Klassische Genmutationen helfen hier nur selten weiter, denn der seltene Tumor weist vergleichsweise wenige Veränderungen im Erbgut auf – im Durchschnitt weniger als eine Mutation pro Million DNA-Bausteine. Das schränkt die Wirkung vieler bekannter Medikamente ein.
Genaktivität und Eiweiße liefern Hinweise für passende Therapien
Statt einzelner Mutationen untersuchten die Forscher, welche Gene im Tumor besonders aktiv sind und welche Eiweiße gebildet werden. Daraus ließen sich konkrete Angriffspunkte für Medikamente ableiten.
Ein Großteil der Therapieempfehlungen beruhte auf diesen Daten. Häufig betraf das Signalwege, die das Wachstum der Krebszellen beeinflussen, sowie Strukturen auf der Zelloberfläche.
Dazu gehörten unter anderem:
- Tyrosinkinasen, die Zellwachstum steuern
- SSTR3 als Ansatz für nuklearmedizinische Behandlungen
- CLDN6 als Ziel für moderne Zelltherapien
- ERBB2, bekannt aus anderen Krebsarten
Medikamente setzen direkt an der Oberfläche der Tumorzellen an
Beim Molekül ERBB2 fiel auf, dass es zwar auf den Tumorzellen vorhanden war, im Zellinneren aber keine starke Aktivität zeigte. Für die Behandlung ist das wichtig.
Medikamente wie Trastuzumab Deruxtecan nutzen diesen Unterschied: Sie binden an die Zelloberfläche und schleusen einen Wirkstoff direkt in die Krebszelle ein. Dabei spielt es keine große Rolle, ob im Inneren ein Signal aktiv ist.
Zwei Patienten erhielten diese Therapie nach mehreren erfolglosen Behandlungen. In beiden Fällen hielt die Wirkung deutlich länger an als üblich.
Patienten profitieren über viele Monate hinweg
Ein anderer Patient blieb über 18 Monate stabil. Bei einer betroffenen Frau blieb die Krankheit mehr als zwei Jahre lang unter Kontrolle. Die Autoren der Studie schreiben hier von „lang anhaltenden Remissionen“.
Solche Verläufe sind bei DSRCT selten, da die Erkrankung normalerweise rasch fortschreitet. Beide Patienten hatten zuvor zahlreiche Therapien hinter sich – einer hatte bereits zwölf verschiedene Behandlungen erhalten.
Auch ein anderes Medikament brachte messbare Effekte. Neun Patienten wurden mit Pazopanib behandelt. Bei fünf von ihnen konnte die Erkrankung kontrolliert werden. In einem Fall hielt dieser Zustand mindestens 17 Monate an.
Individuelle Therapien wirken oft länger als vorherige Behandlungen
Insgesamt erhielten 13 der 30 Patienten eine Therapie, die jeweils auf ihren Weichteiltumor abgestimmt war. Acht von ihnen profitierten davon messbar:
- 5 Patienten zeigten ein Schrumpfen des Tumors
- 3 Patienten hatten einen stabilen Verlauf
Das entspricht einer Remission bei 62 Prozent der behandelten Patienten. Bei sieben Patienten hielt die Wirkung länger an als bei der Behandlung zuvor, was für einen echten Vorteil spricht.
Diese Therapieansätze kommen für seltenen Weichteiltumor infrage
Neben den bereits eingesetzten Behandlungen ergaben sich weitere mögliche Ansätze. 17 von 30 Patienten kamen für klinische Studien infrage. Gerade bei seltenen Tumoren eröffnet das zusätzliche Möglichkeiten.
Im Gespräch sind unter anderem:
- nuklearmedizinische Therapien über SSTR3
- CAR-T-Zelltherapien gegen CLDN6
- Medikamente, die Reparaturmechanismen der DNA blockieren
Im Verlauf der Erkrankung veränderte sich der Tumor bei einigen Patienten. Spätere Proben zeigten mehr genetische Veränderungen als frühere. Wiederholte Analysen können also dabei helfen, die Behandlung anzupassen.
Kurz zusammengefasst:
- Bei einem seltenen Weichteiltumor wird die Diagnose oft zu spät gestellt: In einer Heidelberger Studie wurde sie in 27 Prozent der Fälle erst durch genaue Analysen der Krebszellen korrigiert.
- Individuell abgestimmte Therapien, die auf Eigenschaften der Tumorzellen basieren, zeigten bei 62 Prozent der behandelten Patienten eine messbare Wirkung.
- Besonders auffällig: Einzelne moderne Wirkstoffe konnten das Tumorwachstum über viele Monate bis hin zu mehr als zwei Jahren bremsen.
Übrigens: Während neue Analysen seltene Weichteiltumoren besser verständlich machen, arbeiten Forscher bereits daran, Immunzellen direkt im Körper gegen Krebs umzuprogrammieren. Erste Versuche zeigen, dass Tumoren in wenigen Wochen verschwinden können. Mehr dazu in unserem Artikel.
Bilder: © NCT Heidelberg